|
|
|
|
|
Basic requirements for the XRD sample preparation |
|
|
|
|
|
|
|
|
|
|
The quality of the collected data cannot be superior to the quality of
the sample and to the quality of its preparation and deposition.
That is:
It has absolutely no meaning to lose time of data affected by errors
arbitrarily introduced by the poor care taken in sample preparation.
THE SUCCESS OF A POWDER DIFFRACTION STRUCTURAL
ANALYSIS IS ABSOLUTELY DEPENDENT ON THE NATURE
OF THE SAMPLE AND OF THE EXPERIMENTAL CONDITIONS
Obviously, depending on the final goal of the analysis (phase
identification, polyphasic mixture qualitative and quantitative
analyses, indexing, cell refinement, structural determination,
etc.) the necessary level of “care” can vary and the time and
costs of sample preparation and data collection can be
greatly reduced.
The optimal condition making a sample “suitable” for
diffraction analysis is that its diffraction data are reproducible
on varying the preparation method.
For example, the I/Ic or RIR for the quantitative analyses are
estimated through statistical analysis of different sampling
and measurements.
The majority of the materials examined by XRD initially appears in a
“non-optimal” aggregation state (rocks, minerals, lacquers,
conglomerates, tablets, etc.) and need to be ground to obtain a fine
powder.
How?
Common case: Mortar and pestle (glass for soft materials, agate for
hard materials, BN for extremely hard species.
Or: Mechanical millers with agate, metal alloy, tungsten carbide beads.
Grinding times: seconds to minutes.
Caution! A prolonged grinding may create a large specific surface (with
surface reconstruction or transformation), merging of particles, loss of
crystallinity or even chemical reactions (phase transitions, desolvation,
polymerizations).
 Other tricks
Other tricks:
to make a plastic material fragile, one can perform the grinding or
milling within a liquid nitrogen environment.
repeated heating (10-30 min) of the sample at about 1/3 of its
m.p. (in K) may eliminate defects.
usage of a chemically inert liquid to avoid clustering of soft
particles.
sieving between 25 - 75 µm, also under pressure or within a flux
of an inert liquid.
Caution: Avoid contamination by the mortar or by the miller.
Caution:Problems with ductile materials.
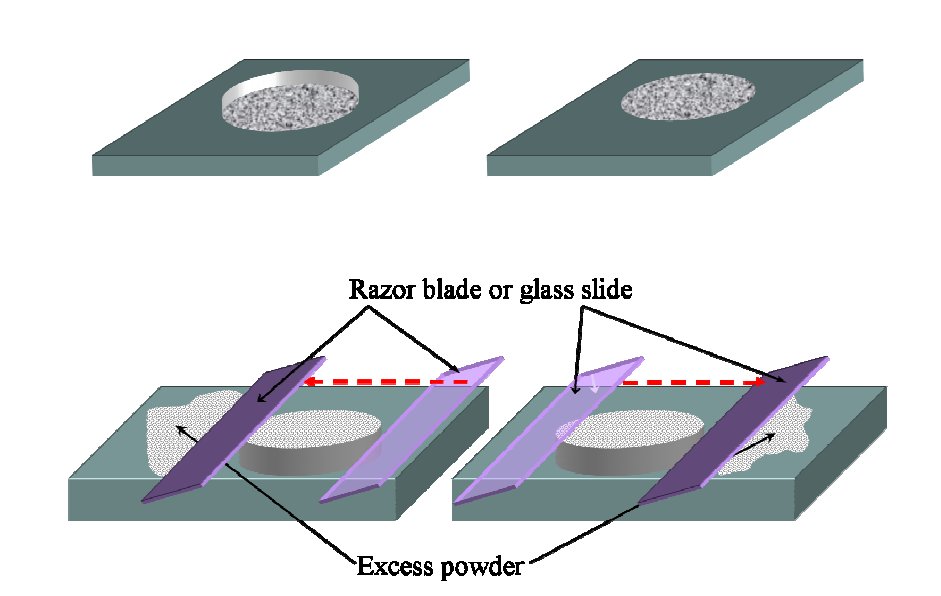
Sample Deposition – Flat Plate (Bragg-Brentano)
plastic, aluminum or glass sample holder: dry sample in hollow space
avoid vertical loading (preferred orientation effects)
Other Methods for Sample Deposition ((Bragg-Brentano)
Dry Dusting
Dusting in oil, grease, silicones, etc.
Back filling or Side Loading
Mixing with inert powder (wheat, cabosil®)
Volatile inert liquids (acetone, ethanol, etc.)
Non volatile suspending inert liquids (amyl acetate, + 5% collodion)
Non volatile suspending inert liquids (amyl acetate, + 5% collodion)
Spray drying (with “spherical” particles)
Thin film (for transparent materials and indexing procedures...)
Note: often it is better to use a zero background plate as sample holder
(Si or SiO2 monocrystal)
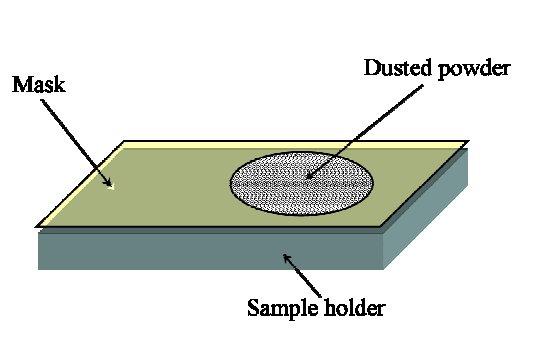
Other Methods for Sample Deposition (Transmission)
Dusting on transparent films, with, or without, lacquers or inert liquids
Dusting on metal (C, Ni) grids
Other Methods for Sample Deposition (Debye-Scherrer)
Capillary soaked into a liquid “ligand” (oils or paraffins) and then in dispersed powders. Mixing of liquid and powders can also precede the deposition.
Capillary loaded by gravity with dry powder and ultrasonic bath
Capillary externally covered by “sticking” powder
Sealed Capillary, with inert gases or mother liquors for unstable samples
Preferential Orientation (or, is some cases, “Texture”)
One of the basic assumptions of meaningful XRD is the homogeneous
spatial and angular, distribution of the crystallite orientations in the
different directions.
Any deviation from homogeneity make sampling of intensities
(the reproducibility of the measurement) rather difficult.
Nevertheless, in a few cases, this effect is informative of the
genesis and microstructure of the sample (deformed metal,
geological sediments, rolling and cleavage, etc.).
Elimination of preferred orientation effects (one of the most
common experimental problems!) would not exist if the crystals
were morphologically spherical.
But the crystal world is intrinsically vectorial, not scalar, and
anisotropy (structural and morphological) in the rule, not the
exception!
Preferred orientation, it not completely avoidable, must be reduced as
much as possible, and later interpreted on the basis of cleavage
properties or on the crystal structure (evidencing, whenever possible,
sections with weak intermolecular contacts).
Are accuracy values 2theta < 0.02° essential?
Estimate of the significance of the results vs. the experimental
accuracy, using numerical analysis of the diffraction peaks location.
What is the best way to determine the TRUE (?) peak positions
How strict is the requirement for high accuracy (not high precision!) ?
Reference: Norberto Masciocchi, Data collection: Experimental set-ups and Sample Preparation, International Workshop on
Structural Determination from Powder Diffraction Data, Villigen (CH), June 18-22 2008.
|
|
|
|
|
|
|
|
|